Department of Cardiology. Cardiology in the Young (CITY group). The Heart Hospital. Londres. Reino Unido.

Las enfermedades del miocardio son afecciones primarias del músculo cardíaco, en su mayoría de origen genético1. La penetrancia dentro de una misma familia y la heterogeneidad, tanto en el modo de presentación como en el pronóstico, hacen que el tratamiento de estas enfermedades requiera a menudo una valoración por parte de centros especializados. El modo de presentación inicial puede ser una muerte súbita (2). La mayoría de los casos de muerte súbita en los países desarrollados se deben a cardiopatía isquémica.
Sin embargo, las causas fundamentales de los casos de muerte súbita en pacientes < 35 años son las enfermedades cardiovasculares hereditarias y las arritmias (3).
Aunque la incidencia varía según las distintas series y edades, se estima que se sitúa alrededor de 0,35-1,28/1.000 habitantes/año2. Dado que la mayoría de los estudios sólo consideran las muertes intrahospitalarias, estas cifras constituyen una apreciación a la baja. El número de estos casos aumenta con la edad y el acmé está en los 60-65 años.
La muerte súbita en jóvenes < 35 años es menos frecuente A menudo, tanto los profesionales de la salud como los gestores de recursos sanitarios consideran el síndrome de muerte súbita en términos numéricos y, como tal, su importancia es relativizada al compararla con otras enfermedades más prevalentes. Sin embargo, la muerte inesperada de una persona joven es un fenómeno devastador, ocurre en la mayoría de los casos en sujetos que, por otro lado, se considerarían sanos y a menudo en individuos devotos de la práctica deportiva.
La incidencia real de muerte súbita en adultos jóvenes está poco documentada en la bibliografía. Además, hay un grupo de muertes en las que el corazón es morfológicamente e histológicamente normal pero que, sin embargo, son muertes por causa arrítmica. La alteración subyacente en estos casos es de nuevo de causa genética como, por ejemplo, en el síndrome del QT largo. Algunas de estas muertes súbitas pueden quedar enmascaradas al atribuirse al consumo de tóxicos.
Entre las causas más frecuentes de muerte súbita están las miocardiopatías, enfermedades primarias del miocardio de origen genético. Las distintas miocardiopatías, miocardiopatía hipertrófica, miocardiopatía dilataday miocardiopatía arritmogénica del ventrículo derecho, entre otras, son causas reconocidas de muerte súbita. El diagnóstico de estas enfermedades en la autopsia tiene obvias implicaciones para los familiares, ya que el patrón de herencia más frecuente es el autosómico dominante. En el momento actual muy pocos sistemas sanitarios han contemplado de forma protocolizada los recursos necesarios para la investigación, identificación y prevención de estos casos. Los factores que contribuyen a este fenómeno son, por un lado, la falta de reconocimiento por la dificultad diagnóstica y, por otro, la falta de provisión de medios.
Es importante destacar iniciativas como la tomada recientemente por el Departamento de Salud del Reino Unido, con la reciente publicación del plan nacional de prevención de muerte súbita (Nacional Service Framework. Chapter 8: Arrhythmias and Sudden Cardiac Death) (3). Por primera vez se reconoce la necesidad de evaluar y derivar a los pacientes y sus familiares para ser valorados en centros donde la atención pueda ser global. Además, se reconoce la responsabilidad del sistema de salud como garante de que tales medidas se hagan efectivas y sean, por lo tanto, guías de lo que se considera buena práctica clínica. Aunque es todavía reciente y no se ha plasmado en una realidad práctica, supone un hito el reconocimiento de que tanto los pacientes como los familiares con historia familiar de muerte súbita y arritmias necesitan tratamiento especializado.
El plan identifica los puntos clave en los que el anatomopatólogo y el médico forense desempeñan un papel fundamental. Por desgracia, éste es el punto de partida para muchas familias. Cuando se identifica un caso, la familia necesita apoyo y también una correcta valoración clínica. Los hallazgos, las muestras de tejido y la disponibilidad de material para un posible estudio genético serán piezas cruciales para la valoración posterior del resto de la familia.
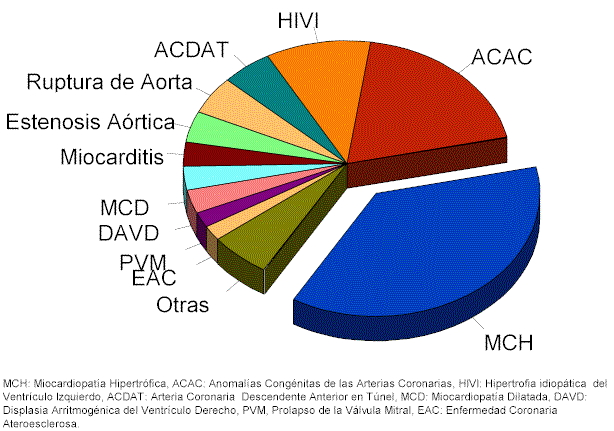
En este punto merece la pena reflexionar sobre el problema epidemiológico que suponen estos síndromes.
Con el desarrollo de la genética, entidades como la hipertrofia ventricular inexplicada pasan a menudo a ser parte del espectro de la miocardiopatía hipertrófica, o bien representan alteraciones mitocondriales o síndromes con hipertrofia ventricular y alteraciones del sistema de la conducción. Entre estos últimos es importante recordar el síndrome de hipertrofia ventricular izquierda asociado con preexcitación basado en alteraciones de los genes que codifican la subunidad γ2 de la proteincinasa dependiente de AMP cíclico (PRKAG2)4. Esta entidad morfológicamente idéntica a la miocardiopatía hipertrófica es, en realidad, una alteración en el metabolismo del glucógeno.
En las últimas 2 décadas se ha avanzado en la identificación de los arcadores de riesgo asociados con muerte súbita en la miocardiopatía hipertrófica. El riesgo anual en series no seleccionadas se estima alrededordel 1%. También se ha estudiado el valor predictivo de los factores de riesgo (5) relacionados con un aumento de riesgo de un evento fatal. Estos factores de riesgo son la presencia de síncope inexplicado, la historia familiar de muerte súbita, la presencia de taquicardiaventricular no sostenida en el Holter, la respuesta plana o hipotensiva de la presión arterial con el ejercicio(6), los antecedentes de parada cardíaca resucitada y la hipertrofia ventricular severa. La presencia de gradiente en el tracto de salida del ventrículo izquierdo, así como el tamaño auricular izquierdo, se relacionan también con un peor pronóstico. Las recomendaciones actuales indican que, en presencia de 2 o más factores de riesgo, se debe recomendar la implantación de un desfibrilador automático (7). En los casos en los que sólo hay un factor de riesgo es muy importante individualizar (8,9).
La presencia de taquicardia ventricular no sostenida es rara en los jóvenes con miocardiopatía hipertrófica, pero cuando aparece se acompaña de un pronóstico ominoso.
La muerte se origina por el desencadenamiento de arritmias ventriculares malignas, ya sea taquicardia ventricular sostenida o fibrilación ventricular, que en estas edades es más frecuente. Los desencadenantes del fenómeno arrítmico fatal son varios y a menudo diferentes para las enfermedades del miocardio que para las enfermedades de los canales iónicos. Es importante, sin embargo, reconocer el patrón y los síntomas relacionados con el evento fatal, como pueden ser el síncope, particularmente si está relacionado con el ejercicio, y las palpitaciones con repercusión hemodinámica.
Éstos deben considerarse de igual importancia que la presentación típica del infarto agudo de miocardio.
La miocardiopatía arritmogénica del ventrículo derecho es una causa reconocida de muerte súbita. En Europa, particularmente en Italia, es la primera causa de muerte súbita en deportistas10. El sustrato patológico se basa en mutaciones de los genes cofidificadores de las proteínas de las uniones intracelulares. El estrés mecánico podría desempeñar un papel fundamental en esta entidad como promotor de apoptosis y sustitución del tejido miocárdico por tejido fibroadiposo(11). Mientras que el estadio en el que la dilatación del ventrículo derecho y las arritmias ventriculares son patentes es relativamente fácil de diagnosticar, la progresión hacia el fallo biventricular o la presentación inicial con cambios sólo microscópicos pueden hacerla indistinguible de casos de miocardiopatía dilatada o corazones con una estructura normal, respectivamente. El diagnóstico en vida es, por lo tanto, difícil y se realiza mediante criterios de probabilidad mayores y menores. Es importante destacar que uno de los criterios mayores es la presencia de antecedentes de muerte súbita con demostración histológica del diagnóstico de miocardiopatía arritmogénica del ventrículo derecho. El diagnóstico genético mejorará de forma indudable la valoración de esta entidad. Aunque no hay series prospectivas que identifiquen los posibles marcadores de riesgo, algunos estudios realizados en pacientes con desfibriladores y miocardiopatía arritmogénica del ventrículo derecho han permitido identificar potenciales marcadores de riesgo (12). Éstos son el síncope, la afección del ventrículo izquierdo, la presencia de arritmias ventriculares sintomáticas y la historia familiar de muerte súbita(12).
Una proporción importante de los casos de muerte súbita puede prevenirse con el diagnóstico precoz y la identificación de los individuos de alto riesgo. La implantación de desfibriladores automáticos de forma profiláctica es eficaz para la prevención de muerte súbita en estos pacientes.
En el presente número de REVISTA ESPAÑOLA DE CARDIOLOGÍA se publica un estudio retrospectivo13 altamente ilustrativo y riguroso de los casos de muerte en una población de niños y jóvenes < 35 años. Esta experiencia es de particular importancia, ya que hay grandes limitaciones en la bibliografía en cuanto a series con correlación anatomoclínica.
Las enfermedades causales son, en su mayoría, enfermedades hereditarias o, al menos, enfermedades en las que hay que sospechar un sustrato genético.
Las miocarditis merecen especial atención, son particularmente frecuentes en la infancia y una de las principales causas de miocardiopatía dilatada. Las causas de muerte relacionadas con las miocarditis agudas o subaguda son la progresión a insuficiencia cardiaca congestiva, los fenómenos tromboembólicos y las arritmias. Es particularmente interesante la observación de los fenómenos embólicos en la serie descrita13. Las indicaciones de anticoagulación permanente o transitoria para la prevención primaria de los fenómenos tromboembólicos en la infancia son escasas y difieren de la práctica o las recomendaciones para enfermedades similares en adultos. Éste es el caso de la dilatación severa del ventrículo izquierdo asociada con disfunción ventricular. Estudios realizados en familias con alta incidencia de miocardiopatía dilatada han demostrado que la presentación temprana a menudo se relaciona con un episodio de infección viral. La teoría infecciosa como desencadenante de la expresión del fenotipo miocardiopatía dilatada se ha demostrado en modelos animales (14).
Aunque hay un patrón de herencia ligado al cromosoma X en algunas miocardiopatías, el patrón de herencia más frecuente es el autosómico dominante. La afección por sexos debería ser, por tanto, proporcional, pero todas las series descritas, incluida la de Morentin et al (13), reflejan un porcentaje mayor de varones.
Las arritmias relacionadas con el ejercicio son motivo de controversia y a menudo las recomendaciones en este sentido no son clínicas, sino médico-legales. Estas recomendaciones varían en los diferentes países. La mayor parte de los casos de muerte súbita en jóvenes no están relacionados con el ejercicio. Sin embargo, hay algunas miocardiopatías en las que el ejercicio podría desempeñar un papel desencadenante principal.
Entre ellas están la miocardiopatía arritmogénica del ventrículo derecho y la miocardiopatía hipertrófica con obstrucción severa. No hay una clara relación entre la miocardiopatía hipertrófica sin obstrucción y con características morfológicas leves y el aumento de riesgo de muerte súbita relacionado con el ejercicio.
Es importante repetir de forma sistemática la valoración de los factores de riesgo, particularmente durante la adolescencia y la juventud, ya que en la mayor parte de los casos los cambios más importantes ocurren en esta etapa de la vida.
La muerte súbita es, por lo tanto, una causa de muerte poco frecuente en jóvenes, pero representa en sí una entidad cuyo impacto social es alto y que precisa investigación multidisciplinaria y adecuación de recursos.
Estos recursos son necesarios para optimizar la certeza diagnóstica y para establecer una adecuada identificación de los individuos con alto riesgo, así como para proponer terapias preventivas.
El estudio forense debe considerarse como piedra angular para la identificación y el punto de partida a la hora de evaluar a una familia afectada. La disponibilidad de tejidos, así como el adecuado consentimiento informado para el posible estudio genético de las muestras, es crítico para cualquier estudio posterior (15).
Para que esto se realice de forma eficaz es importante reunir esfuerzos y actualizar las recomendaciones del enfoque global del síndrome de muerte súbita.
BIBLIOGRAFÍA
1. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, et al. Report of the 1995 World Health Organization/ International Society and Federation of Cardiology Task Force
on the definition and classification of cardiomyopathies. Circulation. 1996;93:841-2.
2. Priori SG, Aliot E, Blomstrom-Lundqvist C, Bossaert L, Breithardt
G, Brugada P, et al. Task Force on Sudden Cardiac Death, European Society of Cardiology. Europace. 2002;4:3-18.
3. National Service Frame Work for Coronary Heart Disease. Chapter eight: arrhythmias and sudden cardiac death. 2005. Disponible en: http://www.dh.gov.uk/assetRoot/04/10/60/40/04106040.pdf
4. Oliveira SM, Ehtisham J, Redwood CS, Ostman-Smith I, Blair EM, Watkins H. Mutation analysis of AMP-activated protein kinase subunits in inherited cardiomyopathies: implications for kinase
function and disease pathogenesis. J Mol Cell Cardiol. 2003; 35:1251-5.
5. Alfonso F. Muerte súbita en la miocardiopatía hipertrófica. RevEsp Cardiol. 1996;49:288-304.
6. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-8.
7. Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on
Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24:1965-91.
8. Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol. 2003;42:873-9.
9. Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, Mc-Kenna WJ. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet. 2001; 357:420-4.
10. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998; 339:364-9.
11. Tome Esteban MT, García-Pinilla JM, McKenna WJ. Actualización en miocardiopatía arritmogénica del ventrículo derecho: genética, diagnóstico, manifestaciones clínicas y estratificación deriesgo. Rev Esp Cardiol. 2004;57:757-67.
12. Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004;109:1503-8.
13. Morentin B, Suárez-Mier MP, Aguilera B, Bodegas A. Mortalidad por enfermedades del miocardio en niños y jóvenes. Estudio observacional de base poblacional. Rev Esp Cardiol. 2006;59: 238-46.
14. Arbustini E, Morbini P, Pilotto A, Gavazzi A, Tavazzi L. Familial dilated cardiomyopathy: from clinical presentation to molecular genetics. Eur Heart J. 2000;21:1825-32.
15. Hughes SE, McKenna WJ. New insights into the pathology of inherited cardiomyopathy. Heart. 2005;91:257-64.


No hay comentarios:
Publicar un comentario